Ende 1998 traten die ersten europäischen Qualitäts- und Sicherheitsanforderungen für In-vitro-Diagnostika, die europäische Richtlinie 98/79/EG (IVDD), in Kraft. Nach fast 20 Jahren wurde im Mai 2017 die neue europäische Verordnung (EU) 2017/746 (IVDR) veröffentlicht, die die IVDD aufhebt und einen universellen Qualitätsstandard festlegt, um neuen medizinischen Technologien besser gerecht zu werden.
Die Liste der Aufgaben, die zu erledigen sind, um sicherzustellen, dass neue und bereits registrierte IVD-Produkte mit den Vorschriften übereinstimmen, scheint jedoch für viele Hersteller von In-vitro-Diagnostika lang und kompliziert zu sein. Außerdem ist der Zeitplan für die Umsetzung nicht kompromisslos.
Aber keine Sorge. Wir haben für Sie eine Schritt-für-Schritt-Anleitung zusammengestellt, mit der Sie die neuen EU-Verordnungen organisiert, rudimentär und risikofrei umsetzen können.
1. Kennen Sie den Zeitplan für den Übergang
Der strenge Umsetzungszeitplan setzt die benannten Stellen, Behörden und Hersteller unter Druck. Da ein Verzug mit den Fristen das Risiko birgt, die Autorität und das Geschäft des Unternehmens zu verlieren, ist es von entscheidender Bedeutung, den Überblick darüber zu behalten, wann die Änderungen umgesetzt werden müssen. Es ist auch wichtig zu wissen, bis wann und unter welchen Bedingungen die IVD-Produkte in Verkehr gebracht und/oder auf dem Markt bereitgestellt werden können.
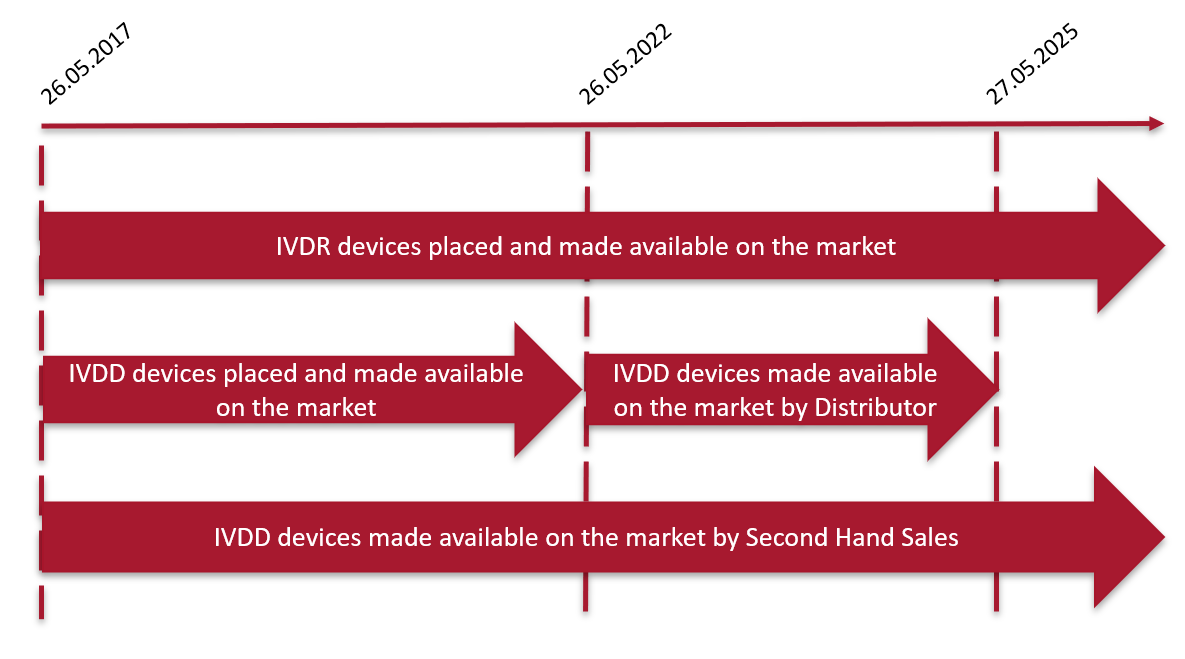
Die wichtigsten Termine in der Übergangszeit der IVD-Verordnung für Produkte, die nicht von einer benannten Stelle zertifiziert werden müssen, sind unten aufgeführt:
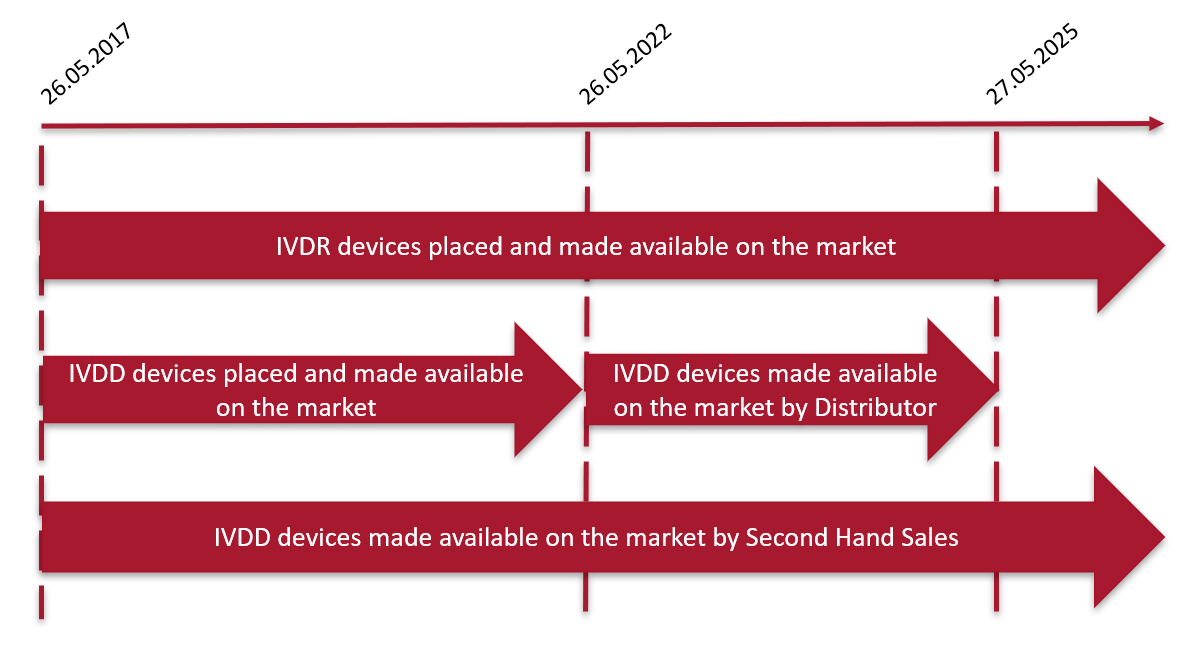
Fristen für IVDD- und IVDR-Medizinprodukte
Basierend auf der assoziierten oder angenommenen Risikoklasse Ihrer Produkte und dem Status der damit verbundenen F&E-Projekte sollte Ihr regulatorischer Fahrplan festgelegt werden, um zu bestimmen, welcher Ansatz am besten zu Ihren Marketingplänen und den bestehenden und neuen Anforderungen der geltenden Richtlinie (IVDD) oder Verordnung (IVDR) passt.
2. Definieren Sie Ihren Anwendungsbereich und Kontext
Ein wichtiger Teil der Regulierungsstrategie besteht darin, den Umfang der Aktivitäten zu berücksichtigen und zu verstehen, die nur für Ihren Betrieb gelten. Berücksichtigen Sie die folgenden Themen:
a) Was ist der definierte oder geplante Verwendungszweck Ihres Produkts?
b) Ist Ihr Unternehmen innerhalb oder außerhalb der EU ansässig?
c) Benötigen Sie eine benannte Stelle zur Bewertung der Konformität Ihres Produkts?
d) Wie sehen Ihre Verkaufs- und Vertriebskanäle aus?
Durch die Beantwortung dieser Fragen können Sie sicherstellen, dass Sie sich auf die für Ihr Unternehmen geeigneten Änderungen der Rechtsvorschriften konzentrieren.
3. Prüfen Sie Ihre Verwendungszweckerklärung sorgfältig
Der Aufwand für die anstehenden Prozesse und die Dokumentation hängt von der Risikoklasse und dem Verwendungszweck Ihres Produkts ab. Die IVD-Vorschriften definieren vier Risikoklassen von A (geringes Risiko) bis D (hohes Risiko), wobei die Produkte auf der Grundlage des damit verbundenen Risikos in Liste A, Liste B und "sonstige Produkte" eingeteilt werden.
Die gleiche Zweckbestimmung eines Produkts kann daher zu unterschiedlichen Einstufungen nach IVDD oder IVDR führen. Dies hat zur Folge, dass für Produkte, die auf der Grundlage der Selbsterklärung des Herstellers in Verkehr gebracht werden, eine benannte Stelle für die Konformitätsbewertung erforderlich sein kann, was einen zusätzlichen Zeit-, Kosten- und Arbeitsaufwand für Ihre Projekte bedeutet.
4. Aktualisieren Sie Ihr internes Qualitätsmanagementsystem
Die neue EU-IVD-Verordnung erweitert die Anforderungen an das Qualitätsmanagementsystem (QMS) des Herstellers, um alle relevanten Aspekte der Qualität von Prozessen, Verfahren und Produkten abzudecken. Je nach Risikoklasse des Produkts kann das QMS Gegenstand regelmäßiger, unangekündigter oder anlassbezogener Audits durch eine benannte Stelle sein, und je besser Sie auf ein Überraschungsaudit vorbereitet sind, desto besser.
Nach der neuen Verordnung sind die Hersteller verpflichtet, eine Person zu benennen, die für die Einhaltung der Vorschriften verantwortlich ist, die über die erforderliche Sachkenntnis auf dem Gebiet der In-vitro-Diagnostika verfügt und die Mindestanforderungen an die Qualifikation erfüllt.
5. Vermeiden Sie unnötige Bürokratie
In Anbetracht der Tatsache, dass die gesetzlichen Anforderungen durch die neue Verordnung gestiegen sind, ist die Wahl des effizientesten und effektivsten Weges, diese zu erfüllen und umzusetzen, ein Schlüsselfaktor für den Erfolg Ihres Unternehmens.
Ein guter Anfang ist eine sorgfältige Analyse Ihrer derzeitigen Strukturen, Projekte und Pläne, um dann einen stringenten Fahrplan für die Einhaltung der Vorschriften zu entwickeln. Wenn Sie sich auf die erforderliche Konformität konzentrieren und den am wenigsten aufwändigen Implementierungsansatz anwenden, sind Sie auf dem sicheren Weg zu einem schnellen Umsatz Ihrer Geräte für den europäischen Markt.
Wenn Sie an einer detaillierten Checkliste für die Umsetzung der EU-IVDR interessiert sind, klicken Sie unten auf den Download:

Dieser Beitrag ist kein Ersatz für die Befolgung der offiziellen Richtlinien der Europäischen Union.